TEMA 3. BIOENERGETICA MITOCONDRIAL
3.1 Hipótesis quimiosmótica y Potencial electroquímico de protón
La teoría quimiosmótica enunciada por Peter Mitchell, explica cómo la energía derivada del transporte de electrones por la cadena de transporte de electrones se utiliza para producir ATP a partir de ADP y Pi. La bomba de protones: el transporte de electrones está acoplado al transporte de H+ a través de la membrana interna mitocondrial desde el espacio intermembranal. Este proceso crea simultáneamente a través de la membrana interna mitocondrial un gradiente eléctrico (con más cargas positivas en el exterior de la membrana que en la matriz mitocondrial) y un gradiente de pH (el exterior de la membrana está a un pH más ácido que el interior). La energía generada por este gradiente es suficiente para realizar la síntesis de ATP.
Y esto representa energía potencial acumulada como: Gradiente de protones= fuerza móvil de protones ("protonmotive force"), y dado que la membrana es básicamente impermeable a los protones, por lo tanto el gradiente no se desarma por una constante re-entrada de los mismos, y teniendo en cuenta que la ATP sintetasa complejo proteico (conocido también como "lollipops", complejo F1, ATPasa mitocondrial) contiene el único canal para la entrada del protón, por lo tanto a medida que los protones pasan por el canal, se produce la siguiente reacción:
ADP + Pi ---> ATP.

Los Protones (indicados por +) entran nuevamente en la matriz mitocondrial a través de los canales que forma el complejo enzimático de la ATP sintetasa. Esta entrada se acopla a la síntesis de ATP a partir de ADP y Fosfato (Pi)

El esquema en la parte superior muestra una mitocondria. En la animación, se observa como los iones H+ se acumulan en el compartimiento mitocondrial externo (espacio intermembrana). En la imagen inferior, se esquematiza lo que le sucede el hidrógeno cedido por el NADH a la cadena de transporte: los electrones son transferidos a lo largo de las proteinas de la cadena, y el protón al espacio intermembrana,donde genera un gradiente. Los protones re-entran pasando por el complejo ATP-sintetasa, generando ATP.
Puntos claves:
1. Los protones son transferidos a través de la membrana, desde la matriz al espacio intermembrana, como resultado del transporte de electrones que se originan cuando el NADH cede un hidrógeno. La continuada producción de esos protones crea un gradiente de protones.
1. La ATP sintetasa es un gran complejo proteico con canales para protones que permiten la re-entrada de los mismos.
2. La síntesis de ATP se produce como resultado de la corriente de protones fluyendo a través de la membrana:
ADP + Pi ---> ATP
ADP + Pi ---> ATP
3.2 Estructura y función de la cadena respiratoria
En esta etapa se oxidan las coenzimas reducidas, el NADH se convierte en NAD+ y el FADH2 en FAD+. Al producirse esta reacción, los átomos de hidrógeno (o electrones equivalentes), son conducidos a través de la cadena respiratoria por un grupo de transportadores de electrones, llamados citocromos. Los citocromos experimentan sucesivas oxidaciones y reducciones (reacciones en las cuales los electrones son transferidos de un dador de electrones a un aceptor).
En consecuencia, en esta etapa final de la respiración, estos electrones de alto nivel energético descienden paso a paso hasta el bajo nivel energético del oxígeno (último aceptor de la cadena), formándose de esta manera agua.
Cabe aclarar que los tres primeros aceptores reciben el H+ y el electrón conjuntamente. En cambio, a partir del cuarto aceptor, sólo se transportan electrones, y los H+ quedan en solución.
Funciones
- Se encarga de transportar electrones. Los electrones se transportan desde moléculas poco oxidantes hasta el oxígeno que es la molécula más oxidante de la cadena. Las moléculas que inician este transporte de electrones son NADH Y FADH, es decir son las moléculas menos oxidantes de la cadena. Una vez que los electrones son entregados al oxígeno, se forma Agua.
- 2. Debido a que la cadena sólo transporta electrones, los protones son bombeados hacia fuera de la mitocondria, lo que crea un gradiente de protones con una carga muy positiva afuera de la mitocondria y una carga muy negativa adentro. Este gradiente obliga a los protones a volver a entrar a la mitocondria y en el paso hacia adentro pasan por una enzima que forma un túnel de protones llamada ATP sintasa que con la fuerza de entrada de los protones, forma ATP.
Balance energético.
La respiración aerobia produce 36 ATP por una molécula de glucosa, sumando los ATP de la glucólisis más los del ciclo de Krebs más los de la Cadena respiratoria.
La respiración aerobia produce 36 ATP por una molécula de glucosa, sumando los ATP de la glucólisis más los del ciclo de Krebs más los de la Cadena respiratoria.

3.3 Fosforilación oxidativa y síntesis de ATP
La transferencia de electrones en la cadena de transporte de electrones es energéticamente favorable porque el NADH es un poderoso donador de electrones y el Oxígeno molecular es un potente aceptor de electrones. De hecho el flujo neto de electrones desde el NADH hasta el Oxígeno resulta en la síntesis de ATP. La fosforilación oxidativa es una serie de eventos químicos que llevan a la síntesis de ATP:
ADP + Pi ® síntesis del ATP
fosforilación del ADP
El evento vital se lleva a cabo en la membrana plasmática bacteriana, en la membrana interna mitocondrial y en los tilacoides de los cloroplastos.
En la década de los 30´s: Belitzer y Tsivakoba encontraron que el proceso de la fosforilación de ADP en los tejidos animales estaba asociado a la respiración o consumo de O2. Mas adelante se describió que la respiración se lleva a cabo en las mitocondrias.
H. Krebs encontró el ciclo de los ácidos tricarboxílicos en el cual el piruvato se transforma en Ac-CoA que a su vez interviene en la reducción de NAD+ y en la posterior generación del succinato.
Como ha sucedido muchas veces a los largo de la historia de la investigación científica, dos investigadores reportaron simultáneamente un evento bioquímico. En 1937, Kalkar en Dinamarca y Belitzer en la antigua URSS, encontraron una correlación muy interesante entre la desaparición del Pi y la respiración. Estudiaron el efecto de la adición de Pi (HPO34) a homogenados de tejidos de mamíferos; el experimento lo realizaron en presencia y ausencia de 02 o en presencia de cianuro (CN-). Reportaron que a medida que se consumía el 02 el Pi desaparecía del medio de reacción y que cuando agregaban a un inhibidor del consumo de 02, CN- e este caso, el proceso no se llevaba a cabo. Posteriormente se verificó que la síntesis de ATP es una reacción endergonica, en la cual la respiración o consumo de 02 acopladas a la fosforilación del ADP, genera energía.
En los seres vivos la oxidación de moléculas orgánicas tiene como resultado el movimiento de protones (H+) del interior de la matriz mitocondrial al espacio intermembranal en mitocondrias y cloroplastos o bien al citoplasma en las bacterias. La cadena de transporte de electrones y la fosforilación oxidativa estuvieron separadas conceptualmente por mucho tiempo. Las observaciones de la formación del ATP hacían pensar a los investigadores en buscaba un intermediario fosforilado de la reación. Hasta que en 1961 Peter Mitchell propuso la hipótesis quimiosmótica en la cual propuso que el intermediario energético necesario para la formación del ATP (o fosforilación del ADP), era una diferencia en la concentración de protones a través de la membrana. Gracias a estas observaciones Mitchell recibió en premio Nobel de Química en 1978. Murió al final de la década de los 80´s.

3.4 Inhibidores y desacoplantes
El uso de inhibidores de la cadena ha permitido trazar el paso de los electrones a través de la cadena y determinar el punto de entrada de diversos sustratos. La velocidad a la cual el oxígeno es consumido por una suspensión de mitocondrias es una medida del funcionamiento de la cadena de transporte de electrones. La velocidad puede ser medida mediante un electrodo de oxígeno.Gran parte del conocimiento de la función mitocondrial ha resultado de estudios con compuestos tóxicos. Inhibidores específicos se han usado para distinguir el sistema de transporte de electrones del sistema de fosforilación oxidativa, y ha ayudado a definir la secuencia de los transportadores redox en la cadena. Si la cadena se bloquea en un punto, todos los transportadores anteriores quedan más reducidos, y los posteriores más oxidados.
Hay seis tipos de venenos que afectan la función mitocondrial:
1. Inhibidores de la cadena que bloquean la cadena respiratoria.
La rotenona, toxina de una planta, utilizada por indios amazónicos como veneno, también ha sido usada como insecticida.
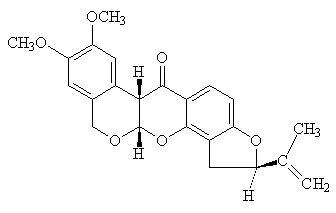
Actúa a inhibiendo el complejo I. Inhibe la reoxidación del NADH, no afecta la del FADH2. Inhibe la oxidación del malato, que es dependiente del NAD+, no así la del succinato. El succinato entra en el segundo punto de entrada a la cadena, posterior al del NAD+.
El amital (barbitúrico) inhibe al complejo I, afecta las oxidaciones dependientes del NAD+.
La antimicina A (Antibiótico).
Actúa a inhibiendo el complejo III. Inhibe la reoxidación del NADH y del FADH2.
El cianuro bloquea el paso de electrones del citocromo a3 al oxígeno.
Estos inhibidores detienen el paso de electrones de modo que no hay bombeo de protones. Sin gradiente de protones, no hay síntesis de ATP.
2. Inhibidores de la fosforilación oxidativa, venenos que inhiben la ATP-sintasa.
La oligomicina, un antibiótico producido por Streptomyces, inhibe a la ATPasa al unirse a la subunidad Fo e interferir en el transporte de H+ a través de Fo, inhibe por lo tanto la síntesis de ATP.
Diciclohexilcarbodiimida (DCCD), un reactivo soluble en lípidos, también inhibe el transporte de protones por Fo al reaccionar con un residuo de glutámico en una de las subunidades de Fo de mamíferos.
En estas condiciones el gradiente de protones que se produce es mayor que lo normal, sin embargo la energía potencial de éste no puede ser utilizada para producir ATP.
3. Venenos que hacen permeable la membrana mitocondrial interna a los protones.
Estos agentes eliminan la relación obligada entre la cadena respiratoria y la fosforilación oxidativa que se observa en mitocondria intacto.Estos venenos, como el 2,4 dinitrofenol (DNP), el carbonilcianuro-p-trifluorometoxi-hidrazona (FCCP) y el carbonilcianuro-m-clorofenilhidrazona (CCCP) desacoplan la fosforilación oxidativa de la cadena respiratoria, se conocen como agentes desacopladores.

Son compuestos liposolubles y ácidos débiles. Las formas disociadas presentan carga negativa altamente deslocalizada, de modo que el campo eléctrico de los aniones es muy débil, ello permite que difundan libremente a través de un medio no polar como las membranas fosfolipídicas. Este comportamiento no es usual, la gran mayoría de iones con carga son excluidos de un ambiente no polar.
La forma protonada, sin carga eléctrica de estos compuestos, pasa a través de la membrana interna mitocondrial intacta, descargando así el gradiente de pH. En la matriz, a pH más bajo, el ácido débil se disocia, la forma disociada pasa la membrana interna, destruyendo el potencial de membrana. Este proceso se puede repetir, de modo que una pequeña cantidad del agente desacoplante puede catalizar el paso de una cantidad enorme de protones y hacer un corto circuito en la cadena respiratoria.
En resumen, permitiendo el paso de protones a través de la membrana, se disipa el gradiente de protones, no hay bombeo de protones a través de la ATP-sintasa con producción de ATP.
Los agentes desacoplantes son todos sintéticos, sin embargo en el mitocondria del tejido adiposo pardo una proteína desacopladora (termogenina) participa en el delicado control de la termogénesis.
4. Inhibidores de transporte (atractalósido) que previenen ya sea la salida del ATP o la entrada de material combustible a través de la membrana mitocondrial interna.

5. Ionósforos (valinomicina, nigericina) que permiten el paso a través de la membrana a compuestos que normalmente están impedidos.
6. Inhibidores del ciclo de Krebs (arsenito) que bloquean una o más enzimas del ciclo de Krebs.
La producción de ATP aeróbica es más eficiente que la producción anaeróbica.
En 1861, Louis Pasteur observó que en levadura expuesta a condiciones aeróbicas, el consumo de glucosa y la producción de etanol decae precipitadamente (Efecto Pasteur).
Glicólisis anaeróbica:
C6H12O6 + 2ADP + 2Pi è 2 lactato + 2H+ + 2ATP
Metabolismo aeróbico de la glucosa:
C6H12O6 + 38ADP + 38Pi 6 O2 è 6 CO2 + 44H20 + 38ATP
El metabolismo aeróbico es más eficiente que la glicólisis anaeróbica en lo que se refiere a producción de ATP.
Sin embargo, como la concentración de enzimas de la glicólisis es alta, de modo que si no están inhibidas, el ATP puede producirse más rápido que a través de la fosforilación oxidativa.
Por otra parte, el cálculo tradicional de 36 ATP o de 38 ATP, según funcione la lanzadera del glicerofosfato o del malato respectivamente, es obsoleto. Mediciones recientes estiman 30 ATP por glucosa totalmente metabolizada.

3.5 Medición del consumo de oxígeno
PROCESOS AEROBICOS
(Glucosa + Oxígeno = Agua + Anhídrido Carbónico + Movimiento y Calor)
Este es un esquema simplificado de las numerosas reacciones que ocurren en el organismo para liberar energía durante el trabajo muscular. Sin embargo, destacan dos aspectos que son fundamentales cuando un trabajador está realizando trabajos físicos pesados. Primero, el "combustible" para el trabajo muscular son los alimentos y estos deben ingerirse en cantidad suficiente y con una distribución adecuada. Segundo, se necesita oxígeno para transformar la energía química contenida en los alimentos en energía mecánica y calórica. El aporte de oxígeno depende de la capacidad de los sistemas respiratorio y cardiovascular, para tomar este elemento desde el aire ambiente y transportarlo hasta los músculos en trabajo.
Una de las metodologías posibles para determinar la respiración celular o mitocondrial es cuantificar el consumo de oxígeno en preparados de células o mitocondrias. Esta cuantificación se puede realizar mediante diferentes técnicas; una de las más sencillas es el electrodo un oxígeno.

ELECTRODO DE OXÍGENO:
El electrodo de oxígeno comprende un cátodo de platino central (B) unido a una resina y un ánodo de plata (C) concéntrico unido por un puente electrolítico y conectados al módulo control. La cámara del electrodo es preparada por aplicación de un espaciador de papel muy fino y una fina membrana de poli-tetra-flúor-etileno (P.T.F.E.) que es cuidadosamente fijada a la placa base donde se encuentran los electrodos por un anillo-O. En la presencia de oxígeno una pequeña corriente fluye a través de los electrodos que es proporcional a la concentración de oxígeno en la muestra. Esta señal es digitalizada por la unidad de control y presentada directamente en el PC.
Estos electrodos pueden ser acondicionados para medidas en fase líquida o en fase gaseosa. Todas las unidades del electrodo deben mantenerse a temperatura constante durante las determinaciones. Este efecto se consigue por circulación de agua a la temperatura deseada alrededor de la cámara y controlando la temperatura de los componentes de la muestra. Este control es importante por dos razones:
1º.- El electrodo es sensible a la temperatura
2º.- El contenido en oxígeno de las muestras acuosas saturadas de aire cambia con la temperatura.
Durante la medida, el electrodo consume una pequeña proporción del oxígeno disponible. Para evitar registrar un declive en la señal debido a este artefacto, las muestras deben estar continuamente en agitación de forma que la capa de líquido, situada encima del disco del electrodo, sea constantemente repuesta en oxígeno.
3.6 Genoma mitocondrial y enfermedades relacionadas
El genoma mitocondrial es una molécula de ADN circular que contiene 16.569 pares de bases y codifica 13 proteínas, 2 ARN ribosomal (ARNr) y 22 ARN de transferencia (ARNt). El código genético que utiliza es degenerado, es decir, ciertos codones en la mitocondria corresponden a aminoácidos diferentes de los utilizados por el genoma nuclear. Sin embargo, depende de muchas proteínas nucleares para poder replicarse y, a su vez, muchas proteínas presentes en las mitocondrias son codificadas por el genoma nuclear.
Las enfermedades mitocondriales son resultado de la falla de las mitocondrias, las cuales son las principales responsables de la creación de la energía del cuerpo necesaria para mantener la vida y apoyar el crecimiento, cuando fallan se genera cada vez menos energía en el interior de la célula, puede entonces presentar lesión celular o incluso la muerte de la célula, así como atrofias o distrofias musculares.

Las mitocondrias son organelas pequeñas, aproximadamente del tamaño de una bacteria, que se encuentran presentes en el citoplasma de las células eucariotas, y cuya función principal es la producción de la mayor parte de la energía celular en forma de ATP. Las etapas finales de esta generación de energía se producen mediante el sistema de fosforilación oxidativa, formado por cinco complejos multienzimáticos localizados en la membrana interna mitocondrial y cuyas subunidades proteicas están codificadas en los dos sistemas genéticos celulares, el nuclear y el mitocondrial. La alteración de la síntesis de ATP puede dar lugar a una serie de trastornos que se conocen genéricamente como enfermedades mitocondriales. Las enfermedades de las mitocondrias parecen ocasionar el mayor daño a las células del cerebro, corazón, hígado, músculo esquelético, riñón, así como sistemas endocrino y respiratorio.
Defectos del metabolismo Mitocondrial
Defectos del metabolismo Mitocondrial
El descubrimiento hace bastante tiempo de la existencia de un genoma mitocondrial abrió un nuevo campo en la patología. La mitocondria es al organela celular donde se obtiene la mayor cantidad de ATP en procesos de oxidación de los distintos substratos: lípidos, acetil coenzima A (proveniente de la decarboxilación oxidativa del piruvato). Hay fosforilación a nivel de substrato en el ciclo de Krebs, pero la vía principal es el acoplamiento de la reoxidación de cofactores vitamínicos reducidos a la síntesis de ATP en la cadena de transporte de electrones. La oxidación de los ácidos grasos a Acetil coenzima A ocurre exclusivamente en la mitocondria. Se sabe desde hace mucho que en algunas enfermedades musculares hay acumulación de material lipídico en las fibras musculares de los pacientes afectados. Actualmente se considera que esto responde a la deficiencia del transporte de residuos de ácidos grasos por baja disponibilidad de carnitina o por fallas en las palmitoil transferasas I o II, aunque no se ha descartado la posibilidad que haya otras enfermedades con acumulos lipídicos que tengan una causa distinta a la deficiencia del sistema de transporte de lípidos al interior de la mitocondria. También se han descrito deficiencias del transporte de los otros substratos para la oxidación mitocondrial, como el ingreso de ácido pirúvico (monocarboxilato translocasa) o su utilización (piruvato deshidrogenasa, piruvato carboxilasa). La beta oxidación de los ácidos grasos también puede estar alterada.
Defectos en la cadena respiratoria
La cadena respiratoria está constituida por al menos 67 péptidos, y entre 11 y 13 de ellos son codificados por el ADN mitocondrial (ADNmt). La mitocondria tiene sistemas de traducción y transcripción propios, pero la gran mayoría de los constituyentes de la cadena respiratoria se sintetizan fuera de la mitocondria y deben ser incorporados a ella por sistemas de transporte complejos. La transmisión de las alteraciones se ve complicada por la posibilidad de herencia maternal, no mendeliana de ADNmt, y por la alta velocidad de replicación y mutación del cromosoma mitocondrial. De un modo muy general, la cadena respiratoria consta de 4 complejos, de los cuales hay dos dispuestos en paralelo (complejos I y II) y dos en serie, tanto entre sí como con los complejos I y II (complejos III y IV). Desde un punto de vista estrictamente fisiopatológico, existen 4 mecanismos posibles de daño:
1. Alteración del grupo prostético (en la enfermedad de Menke hay una concentración de cobre muy disminuida, y la actividad de la citocromo c oxidasa está muy bajo lo normal en el cerebro y posiblemente otros tejidos)
2. Alteración de la traducción - transcripción del ADNmt (aunque no se han descrito patologías por esta causa, la herencia maternal en algunas encefalomiopatías hace sospechar su existencia)
3. Alteración de la traducción-transcripción de ADN nuclear.
4. Alteración del procesamiento post traslacional de las subunidades proteicas (las deficiencias de: sistemas de acoplamiento de los precursores de las subunidades proteicas con receptores de la membrana celular externa, la traslocación de este complejo, el ensamblaje de las subunidades en la membrana mitocondrial interna, conducirían a deficiencia enzimática).
A continuación se describen las patologías más conocidas de los distintos complejos de la cadena de transporte de electrones. En algunos pacientes en estudio se han encontrado enfermedades donde el cuadro era predominantemente muscular, con intolerancia al ejercicio y debilidad. El comienzo era en la infancia o inicios de la adolescencia. Otros casos correspondían a una encefalomiopatía, con enfermedades tales como:
a) Demencia: Enfermedad del Sistema Nervioso Central que se caracteriza por la aparición de múltiples síntomas o síndromes debido a la pérdida de las funciones superiores del Sistema Nervioso Central, aparecen síntomas tales como: alteraciones en la memoria inmediata y retrógrada, orientación, lenguaje, escritura, cálculo, alteración del pensamiento, capacidad de ejecución). Este déficit está determinado bien por la disfunción neuronal, o por la muerte de estás neuronas. Esta enfermedad o pérdida global de funciones tiene un curso progresivo e interfiere en las actividades de la persona, así como en su relación social y laboral.
b) Atrofia óptica: La atrofia del nervio óptico es una incapacidad permanente de la vista causada por daños al nervio óptico. La atrofia puede variar desde parcial, cuando algunos de los axones en la fibra nerviosa se encuentran dañados, hasta profunda, cuando lo están la mayoría. Puede afectar a un ojo o a los dos y también puede ser progresiva, dependiendo de la causa. Las áreas del ojo más vulnerables son las correspondientes a la zona central de la retina, la zona responsable de los detalles y el color (mácula).
c) Miopatía mitocondrial: es decir enfermedades del músculo mitocondrial, infantil fatal familiar, con compromiso muscular y hepático. El componente mitocondrial alterado es la citocromo c oxidasa, con ausencia de los citocromos a3 y b. Un primo de las afectadas mostró también deficiencia mitocondrial hepática. La miopatía mitocondrial infantil benigna parece corresponder a una deficiencia transitoria de la citocromo c oxidasa.
d) En la enfermedad de Menkes (tricopoliodistrofia) se encuentra un desorden recesivo ligado al cromosoma X, que se caracteriza por crisis de comienzo temprano en la infancia, regresión del desarrollo, anormalidades del pelo, arterias tortuosas, huesos frágiles, hipopigmentación (defectos oculares) e inestabilidad de la temperatura. Los afectados fallecen antes de los tres años de edad. Los hallazgos bioquímicos incluyen niveles plasmáticos deprimidos de cobre y ceruloplasmina, que se han atribuido a un transporte intestinal deficiente de cobre. Algunos síntomas podrían corresponder a deficiencias secundarias de las cuproenzimas, tales como la citocromo c oxidasa.
e) Se ha informado de pacientes con oftalmoplegia externa progresiva y miopatía mitocondrial, que presentan ausencia total de citocromo c oxidasa a la unión de anticuerpos específicos.
f) La encefalomiopatía necrotizante sub aguda o enfermedad de Leigh se caracteriza por anormalidades respiratorias, grito débil, dificultades para alimentarse, visión y audición defectuosas, ataxia (inhabilidad de controlar voluntariamente los movimientos musculares, degeneración del cerebelo y otras partes del sistema nervioso), debilidad, deterioro intelectual y crisis convulsivas. El comienzo es en la infancia temprana, aunque algunos afectados se desarrollan normalmente por algunos meses, la muerte ocurre a los pocos años de edad. La autopsia revela lesiones simétricas bilaterales focales desde el tálamo al puente, que comprometen las olivas inferiores y las columnas posteriores de la médula espinal.
g) En la enfermedad de Alper (poliodistrofia progresiva esclerosante) también se ha descrito deficiencia de la citocromo c oxidasa. Hay evidencia de patologías que comprometen múltiples citocromos, y que aparecen ligadas al cromosoma X. Así, un caso de miopatía mitocondrial, con cardiopatía y neutropenia cursaba con defectos de los citocromos c1, c, b y aa3.
Es pertinente comentar que las patologías mitocondriales con compromiso predominante de la cadena de transporte de electrones, que afectan el músculo tanto como al sistema nervioso central.
Para comprender cuáles son las causas de trastornos asociados don defectos en la mitocondria, primero debemos comprender que es lo que ocurre en el interior de la mitocondria.
Enfermedades Asociadas con mutaciones en el ADN Mitocondrial
Como las proteínas componentes de los complejos multienzimáticos de la cadena respiratoria y fosforilación oxidativa están codificadas tanto en el ADN nuclear como en el mitocondrial, estas enfermedades pueden estar causadas por mutaciones en los genes de ambos sistemas genéticos. Sin embargo, habitualmente se conoce con el nombre de enfermedades mitocondriales a los daños producidos en el ADNmt, que presentan un tipo de herencia materna. Las manifestaciones clínicas de estas enfermedades son muy variadas, entre los mas comunes están: demencia, desórdenes motores, intolerancia al ejercicio, accidentes cerebro-vasculares, convulsiones, oftalmoplegia, retinopatía pigmentaria (severa disminución de la agudeza visual y en muchas ocasiones conlleva a la ceguera, esta enfermedad es causada por la degradación de la retina), atrofia óptica, ceguera, sordera, cardiomiopatía, disfunciones hepáticas y pancreáticas, diabetes, falta de crecimiento, anemia sideroblástica, pseudo obstrucción intestinal, nefropatías (movilidad anormal del riñón), estatura corta, acidosis metabólica y otros más secundarios.
En general, son trastornos multisistémicos que afectan fundamentalmente a los tejidos y órganos que mas dependen de la energía mitocondrial (sistema nervioso central, músculo cardiaco y esquelético, riñones y sistema endocrino). Sin embargo, al estar las mitocondrias presentes en todos los tejidos, otros muchos órganos pueden estar implicados en estos síndromes tan heterogéneos. De hecho, una de las pistas que conduce a la sospecha de enfermedad mitocondrial es la implicación de muchos órganos diferentes. Otros caracteres morfológicos y bioquímicos que suelen estar asociados a enfermedades mitocondriales son la presencia de fibras rojo-rasgadas (acumulación de mitocondrias anormales en tamaño y número) en biopsias musculares teñidas con tricromo de Gomori, la presencia de fibras no reactivas a la tinción histoquímica de la citocromo c oxidasa, y defectos en uno o varios complejos de la cadena respiratoria. Sin embargo, algunas enfermedades claramente mitocondriales no presentan estos caracteres tan típicos de los trastornos mitocondriales, especialmente en pacientes en edad pediatrica. Dada la variedad de las manifestaciones clínicas, morfológicas y bioquímicas presentes en las enfermedades mitocondriales, su clasificación se basa en las características moleculares y genéticas de las mutaciones.

{kind=link}
Así, las enfermedades mitocondriales se pueden dividir en dos grandes grupos: Enfermedades asociadas a mutaciones puntuales y enfermedades debidas a reorganizaciones del ADNmt. A continuación se hace una relación de las enfermedades comunes asociadas a estos tipos de mutaciones.
Neuropatía óptica hereditaria de Leber.
La neuropatía óptica hereditaria de Leber (LHON) fue la primera enfermedad humana heredada por vía materna que se asoció con una mutación en el ADNmt. Está caracterizada por una ceguera bilateral aguda o subaguda, originada por atrofia del nervio óptico, que aparece en la segunda o tercera década de la vida y que afecta más a hombres que a mujeres. Normalmente, los pacientes solo tienen afectada la visión, pero en algunos casos puede ir acompañada de anormalidades en la conducción cardiaca, ataxia cerebelar, neuropatía periférica que puede ser:
* Sensorial: pérdida de protección
* Motora: pérdida del tono muscular, atrofia, deformidades.
* Autonómica: ausencia de suduración = fisuras.
Son muchas las mutaciones puntuales que se han asociado con esta enfermedad, pero solamente 3 de ellas, las localizadas en los nucleótidos 11.778, 3.460 y 14.484. Todas estas mutaciones están localizadas en genes estructurales.La mayor predominancia de la enfermedad en hombres sugiere que puede existir alguna influencia de un gene nuclear situado en el cromosoma X, y de hecho, se han encontrado muchas de estás enfermedades en la población filandesa.
Síndrome de neuropatía, ataxia y retinitiopatía pigmentaria (NARP).
Este síndrome, caracterizado por una debilidad muscular, retraso en el desarrollo, neuropatía sensorial (pérdida de protección), convulsiones, ataxia, demencia y retinopatía pigmentaria (severa disminución de la agudeza visual) y se ha asociado a un cambio Timina Guanina en el nucleótido 8.993 de la subunidad 6 de la ATPasa.
Síndrome de Leigh de herencia materna (MILS)
El síndrome de Leigh es una enfermedad muy variada con trastornos degenerativos en muchas partes del organismo, que aparece en el primer año de vida. Es una enfermedad muy devastadora que se caracteriza por disfunciones del tallo cerebral y de los ganglios basales, desmielinización, regresión psicomotora, retraso en el desarrollo, convulsiones, ataxia, neuropatía periférica. La presencia de lesiones necróticas cerebrales focales en el tálamo, tallo cerebral y núcleo dentado confirman el diagnóstico. Es producida por la mutación Timina Guanina en el nucleótido 8.993, pero con un porcentaje de la mutación por encima del 90%. Formas menos severas de esta enfermedad se han asociado con un cambio en la misma posición del ADNmt. Asimismo, se ha descrito por primera vez una mutación en el gene nuclear de la subunidad flavoproteica de la succinato deshidrogenasa del complejo II que causa un defecto en la cadena respiratoria mitocondrial. Esta mutación produce un cambio Citosina Timina en el nucleótido 1684. Este síndrome puede estar causado también por mutaciones en otros genes nucleares que codifican subunidades de la piruvato deshidrogenasa.
Síndrome de encefalomiopatía mitocondrial con acidosis láctica y episodios de accidentes cerebro-vasculares (MELAS).
Este síndrome se ha asociado en el 90% de los casos con una mutación (Adenina Guanina) en la posición 3.243 del genoma mitocondrial. MELAS está caracterizado fundamentalmente por accidentes cerebro-vasculares que provocan una disfunción cerebral subaguda y cambios en la estructura cerebral, acidosis láctica y/o presencia de fibras rojo-rasgadas. Estos caracteres pueden ir acompañados también de encefalomiopatía (trastorno del encéfalo, encargado de la regulación, coordinación y control de todos los procesos orgánicos y de la dirección de las relaciones con el mundo exterior) con convulsiones generalizadas, dolor de cabeza, sordera y demencia. La mutación principal, en la posición 3.243, se ha relacionado también con otras enfermedades muy distintas como oftalmoplegia progresiva externa, cardiomiopatías e incluso con diabetes y sordera, por lo que la relación genotipo-fenotipo no es muy fija.
Síndrome de epilepsia mioclónica con fibras rojo-rasgadas (MERRF)
MERRF es un síndrome de herencia materna caracterizado por epilepsia, debilidad muscular, ataxia, convulsiones generalizadas y miopatía mitocondrial con presencia de fibras rojo-rasgadas. Otros síntomas menos comunes son demencia, sordera, neuropatía, atrofia óptica, fallo respiratorio y cardiomiopatía. Puede aparecer tanto en la infancia como en edad adulta y es de curso progresivo. El 80-90% de los casos de MERRF están asociados con la presencia de una mutación Adenina Guanina en la posición 8.344 del gen del ARN de la mitocondria. Recientemente, se ha demostrado utilizando híbridos transmitocondriales (células r0 repobladas con mitocondrias con la mutación 8.344) que la mutación produce una disminución de la síntesis de proteínas mitocondriales.
Otras enfermedades asociadas a mutaciones puntuales en el ADNmt.
Además de las mutaciones puntuales descritas anteriormente, se ha encontrado un buen número de mutaciones puntuales asociadas a otros muchos síndromes. Entre ellos, la diabetes de herencia materna con sordera que afecta a 15% de la población diabética, las cardiomiopatías de herencia materna; LHON y distonía , la sordera de herencia materna; anemia; deficiencia fatal de la cadena respiratoria infantil, entre otras. Se está estudiando el papel que puede jugar el daño en el ADNmt en enfermedades neurodegenerativas como Parkinson y Alzheimer. Asimismo, se está investigando la posible relación de la inefabilidad masculina con daños en el ADNmt debido a la dependencia que tienen las reacciones de movilidad de los espermatozoides de la función de la cadena respiratoria.
Enfermedades asociadas a reorganizaciones del ADNmt
Algunos pacientes con enfermedades que afectan al sistema de fosforilación oxidativa presentan mutaciones originadas por reorganizaciones del ADNmt. Este tipo de mutaciones, al contrario que las mutaciones puntuales, suelen ser espontáneas aunque hay descrito algún caso de herencia materna. Hasta el momento se han descrito varios cientos de reorganizaciones del ADNmt. Son heteroplásmicas y pueden aumentar la gravedad con la edad. Se han descrito una amplia variedad de síndromes clínicos, algunos de los cuales se describen a continuación.
Síndromes de oftalmoplegia externa progresiva (PEO).
La oftalmoplegia externa progresiva crónica está caracterizada por oftalmoplegia, caída de los párpados y enfermedades musculares. Además, suele ir acompañada de intolerancia al ejercicio y debilidad muscular. En general, es una enfermedad benigna que suele aparecer en la adolescencia o en adultos jóvenes. Aparece de forma esporádica sin historia familiar.
Síndrome de Kearns-Sayre.
El síndrome de Kearns-Sayre es una enfermedad multisistémica progresiva que aparece antes de los 20 años de edad y que está caracterizada clínicamente por PEO y retinopatía pigmentaria. Además suele ir acompañada de otros síntomas como ataxia, defectos musculares de la mitocondria, bloqueo de la conducción cardiaca, elevados niveles de proteína CSF (fluido cerebro espinal) por encima de 100 mg/dl, sordera y demencia, fallos endocrinos y renales.
Síndrome de Pearson.
El síndrome de médula ósea-páncreas de Pearson es una enfermedad de los primeros años de vida que afecta al conjunto de fenómenos que conducen a la formación y maduración de los elementos que componen la sangre (hematopoyesis) y a la función pancreática exocrina. Los niños afectados suelen morir antes de los 3 años y los que sobreviven suelen desarrollar mas tarde un fenotipo de Kearns-Sayre. Estos pacientes presentan delecciones grandes únicas del ADNmt, en general son esporádicas aunque se ha descrito algún caso de herencia materna. Estos tres síndromes, PEO, Kearns-Sayre y Pearson, tienen en común el presentar grandes delecciones en el ADNmt que suelen aparecer de forma espontánea. En general son delecciones únicas pero también se han descrito delecciones múltiples. En general, están localizadas en el arco grande comprendido entre los orígenes de replicación e incluyen varios genes del ADNmt. La presencia de estas delecciones se detecta con relativa facilidad, son siempre heteroplásmicas, pueden llegar a constituir un porcentaje muy alto de la población de ADNmt y se encuentran en varios tejidos. Su determinación se hace fundamentalmente en biopsias de músculo pero, si la afectación es muy grave, se pueden llagar a detectar también en sangre. Existen otras muchas enfermedades que están asociadas a la presencia de grandes delecciones en el ADNmt. Entre otras se puede mencionar los fenotipos de diabetes, sordera y atrofia óptica; miopatías en general; el síndrome de encefalomiopatía mitocondrial neurogastrointestinal (MNGIE); diabetes mellitus, diabetes insípida, atrofia óptica y sordera (DIDMOAD). Asimismo, existen síndromes que presentan grandes delecciones en el ADNmt que se trasmiten de forma autosómica dominante o recesiva. Esta forma de herencia sugiere la existencia de genes nucleares que pueden afectar a la estructura del ADNmt.
Alteraciones de la cadena respiratoria mitocondrial
Los defectos de los complejos de la cadena respiratoria mitocondrial tienen una presentación clínica muy variada, que puede presentarse tanto en el neonato como en la edad adulta. El inicio de la síntomatología puede incluso producirse en la embriogénesis ocasionando malformaciones congénitas. La afección de estas enfermedades generalmente es multiorgánica siendo esta una de sus principales características clínicas. Sin embargo, existen una serie de trastornos que deben hacer sospechar una enfermedad mitocondrial. Uno de los más comunes es la oftalmoplejía externa progresiva (OEP) donde los pacientes pueden presentar además trastornos cardíacos, retinitis pigmenteria, sordera, afectación del sistema nervioso central y diabetes. Otros pacientes tienen síntomas que afectan predominantemente al músculo esquelético. Se trata de niños con marcada hipotonía en el periodo neonatal, adolescentes con fatigabilidad muscular, episodios de mioglobinuria o con miopatía. El tercer grupo de pacientes presentan síntomas que afectan fundamentalmente el sistema nervioso central. Se trata de enfermos que sufren un deterioro progresivo de sus funciones superiores con accidentes vasculares cerebrales y acidosis láctica (MELAS) o con epilespsia mioclónica (MERRF). También se han identificado como enfermedades mitocondriales a pacientes con atrofia óptica hereditaria de Leber (LHON). Un signo diagnóstico común para muchos de estos síndromes es la presencia de fibras rojo-rotas en la biopsia muscular (ragged red). Las enfermedades mitocondriales son ocasionadas por alteraciones del ADN nuclear o mitocondrial.
BILIOGRAFIA
{kind=link}
http://www.bioquimicaqui11601.ucv.cl/unidades/cte/traselectfofox4fid.html. BIOENERGETICA MITOCONDRIAL
Debido al alto índice de mutación del ADNmt es posible encontrar un gran número de mutaciones puntuales. Sin embargo, la mayor parte de éstas van a ser mutaciones silenciosas que no van a causar ningún tipo de defecto. Para que una mutación pueda ser considerada como patológica se requiere que cumpla los criterios: que se encuentre en familias afectadas de poblaciones étnicas diferentes, que exista una correlación entre el porcentaje de la mutación y el fenotipo, que segregue junto con el fenotipo y que afecte a una base muy conservada evolutivamente. Se han encontrado más de 50 mutaciones puntuales que se localizan en los tres tipos de genes codificados en el ADNmt.
No hay comentarios:
Publicar un comentario